class: middle center # Organization .footnote.left.title[http://www.riffomonas.org/reproducible_research/organization/] .footnote.left.gray[Press 'h' to open the help menu for interacting with the slides] ??? Welcome back to the Riffomonas Reproducible Research Tutorial Series. Today's tutorial is going to focus on how we can organize our projects to maximize the ability to find files as well as to improve the reproducibility of our analyses. As we do this, we'll practice using Amazon's EC2 service, we'll develop our command line skills, and we'll use Markdown to improve the documentation. As a foretaste for the next tutorial, we'll begin to use version control. I think that many of us have a habit of organizing our projects by having multiple directories and sometimes these directories might be strewn across your hard drive or even on different computers. Alternatively, there's also a temptation to have a single directory where you dump all your raw data, your process data, your code, your figures, your images, and your text. In short, you really have no organization. As you can appreciate, this makes it really hard to find anything and to know where you are in your data analysis workflow. But hopefully, by now you've been able to read William Noble's <i>A Quick Guide to Organizing Computational</i><i>Biology Projects</i>. If you haven't, stop the video now and go back and read that, it's really important. You can find a link for it on the Reproducible Research homepage within the riffomonas.org website. We'll be adapting the structure that Noble describes in that paper for analysis that we'll be working on for the rest of the tutorial series and also, a lot of the ideas that he describes in that paper will be the motivation for future tutorials within this series. This tutorial will have a lot going on in it, feel free to take it in chunks. You'll start to notice that some of the tools we've already discussed, things like Markdown and using AWS, will be prominent and so we'll get extra practice here. You'll also notice that other tools, things like Git and scripting, are introduced but perhaps not dealt with in great depth. You'll become a Git expert in the next tutorial and a scripting expert in the tutorial after that. By seeing the material multiple times and then layering new information on each time and seeing the tools in different contexts, you'll learn the material that much better. So if you feel a bit lost, that's fine, stick with it, it's a part of the program, it's there to help you learn better. Join me now in opening the slides for today's tutorial which you can find within the Reproducible Research Tutorial Series at the riffomonas.org website. --- ## Exercise Can you restart and log into the EC2 instance we created in the fourth tutorial on using high performance computers? ??? Before we get going on today's tutorial on project organization, I'd like you all to do a little exercise with me or for me that will be useful and pretty critical for subsequent steps within this tutorial. So can you restart and log into the EC2 Instance that we created in the fourth tutorial where we discussed using high-performance computers? So go ahead and pause this video and when we come back, I'll show you how I go about logging into the instance. Okay, so did you get it? Let me show you what I would do. So I am going to open up a new tab here in my browser and the web address is aws.amazon.com. I'm going to sign into my console, I'm going to give it my email address and my password and then I'll sign in. It's got in here my recently visited services of EC2, again, you could type EC2 in here but I'll go ahead and press EC2. And I then will click on the link for "Running Instances," and so it says I currently have zero Running Instances because we stopped the Instance before. And so you'll see my Instance here and it says, "The Instance state is stopped," so I want to go ahead and go, "Actions," "Instance State," "Start." "Are you sure?" Yes. And so this might take a couple of seconds to fire up and what we're looking for is a Public DNS address down here or Public IP address. And so if we're a little antsy, we can hit the Refresh button and voilà, there it is. So now I want to open up my terminal and, again, if you're using Windows, then you're probably going to be using that Git Bash...you're going to use the Git Bash terminal window. And so here, remember, I can type ssh -i ~/.ssh/MyKeyPair.pem ubuntu@ and then I'm going to copy and paste over here so hopefully, this works. "Are you sure?" Yes. And ha-ha, we're connected. So it says there's six packages that can be updated, I'm not going to worry about those for now. If I type "ls," I see that 300.jpeg file that's there and if you were doing some other exercises to practice moving things around, you might see other things there as well. I'm going to go ahead and delete that file, so I'll do rm 300.jpeg, okay? So, great, this is how we go about logging into our EC2 Instance. Now, do you remember what that other command was that we could use if we're afraid that our network connectivity is going to break while we're running something? Do you remember what that was called? Right, that is called tmux, so T-M-U-X, and this brings up the tmux session and we know that it's a tmux session because at the bottom of the screen, there's this green bar. Great, so we're going to leave this here, I'm going to leave the instance running, again, on a per-minute or per-hour basis, it's not that expensive and it's probably more expensive to keep, you know, just in terms of our time, to log out and log in over and over again, just we'll leave it open, okay? And so we'll go back to our slides now. And so, hopefully, this exercise was something that you remember, perhaps you wrote down a note to yourself on how to do that, and again, by doing it numerous times over the course of this tutorial series, hopefully, you'll get better adjusted to doing it and it'll just feel like second nature to you. --- ## Learning goals * Evaluate different organizational strategies for their ability to foster reproducibility * Apply `bash` commands to fix a chaotic project so it has a more logical structure * Interface with a project template to improve the reproducibility of your research * Apply material from the documentation and organization tutorials to start a new project * Demonstrate how to use git and GitHub to make your new project public ??? So the goals of today's tutorial are to evaluate different organizational strategies for their ability to foster reproducibility. We're going to use Bash commands to take a fairly chaotic project and give it a more logical structure. We're going to use a project template that will help us to organize our project and hopefully, improve the reproducibility, and we're going to use what we talked about in the documentation tutorial as well as this tutorial to start a new project, right? So there's one thing to retrofit an old project but it's much easier to start fresh from a new project incorporating these ideas. And then finally, we're going to demonstrate how we can use Git and GitHub to make our new project public. So as I said in the introduction, before we go any further, it's really important that you read this article. William Noble did a great job of laying out a lot of really solid principles and recommendations on how we can organize our projects and not only organization but it also talks about things like documentation and automation, things that we'll come back to in future tutorials. --- ## Before we go any further... You need to read: [Noble 2009 *PLoS Comp Biol*](http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000424) ??? So if you've gotten this far and you haven't been listening to me, stop, go back, and listen to this or read this paper. So what did William Noble have to say? So, as I said, it discusses far more than organization, he talks about automation, he talks about how project organization is a form of documentation that helps make written and scripted documentation easier to maintain. --- ## Noble paper on file organization * Discusses far more than organization * Project organization is a form of documentation and helps make written and scripted documentation easier to maintain * Provides an outline for the rest of these tutorials * Organization * Literate programming * Scripting * Automation * Version control .footnote[[Noble 2009 *PLoS Comp Biol*](http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000424)] ??? If you know where things are, then it's easier to maintain your project and to know where things are. If you've got a directory that has your reference files, it's very easy to see which reference files you're using, whereas if all those reference files are scattered into a big garbage can of data in a single directory, then it's very easy to get lost and to not know what files you're using. And as I said, this is going to help provide an outline for the rest of these tutorials and talking about things like organization, literate programming, scripting, automation, version control, and so we'll come back to this paper frequently. So for this tutorial, I want to look at this figure that was in the Noble article and we're not going to use this exact structure but it really helps us to think about how we might want to organize our own projects. --- .center[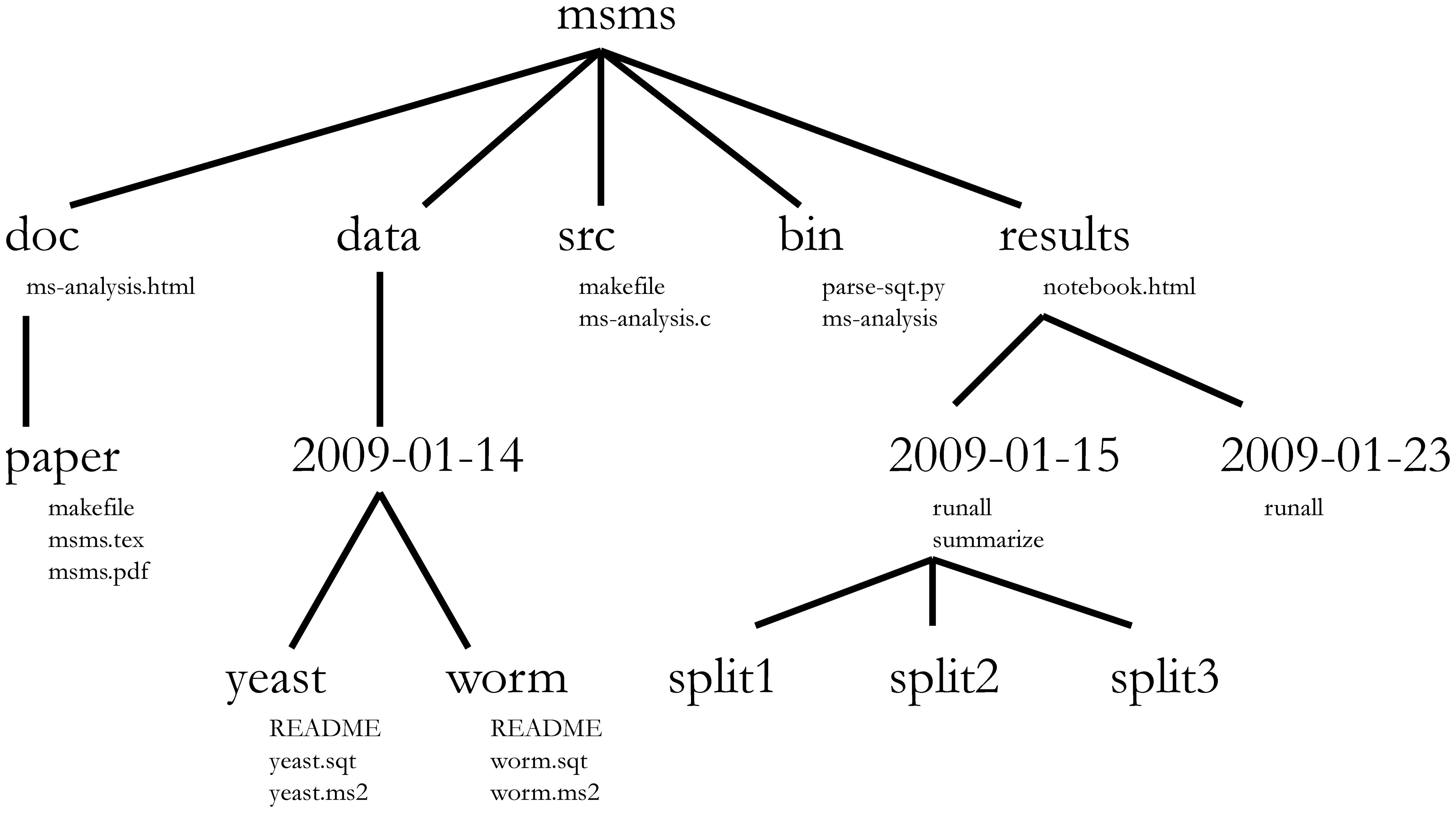] > The core guiding principle is simple: Someone unfamiliar with your project should be able to look at your computer files and understand in detail what you did and why ... Most commonly, however, that "someone" is you. .footnote[[Noble 2009 *PLoS Comp Biol*](http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000424)] ??? And as he says, the core guiding principle is simple: someone unfamiliar with your project should be able to look at your computer files and understand in detail what you did and why. Most commonly, however, that someone is you, okay? And so, if you wanted to find the worm data for this project, it's pretty clear looking at this structure where it is, right? And so you might imagine they're collecting data on different days, and so there might be other directories within that data directory and then for each day, there might be data on yeast and worm or other model organisms, right? And so, again, we're not going to use this exact structure but it's helpful in thinking about the principles we talked about in the previous tutorial of having directory for our documentation, for our manuscripts, for our data, our raw and our processed data, to have a directory for our source code, and perhaps a directory for our results. --- ## Refresher on documentation from Noble * "Record every operation that you perform" * "Comment generously" * "~~Avoid~~[DO NOT] editing intermediate files by hand" * Use a driver script to centralize all processing and analysis * "Use relative pathways [relative to the project root]" * "Make the script restartable" .footnote[[Noble 2009 *PLoS Comp Biol*](http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1000424)] ??? And so some of the big bullet points in the Noble article were to record every operation that you perform, comment generously. He says, "Avoid editing intermediate files by hand," I would say, "Do not edit intermediate files by hand," that we should be using scripts to do that and we should be keeping our raw data raw. We should develop a driver script to centralize all processing and analysis, and then we should use relative paths, relative to the project root, and then we should make the script restartable. So, again, we're going to...we've already talked about commenting generously, we've talked about keeping raw data raw, and so here we're going to talk about one or two of these other bullet points and in subsequent tutorials, we'll take on the others. --- ## Compare file organization from two projects * Examples * [Meadow et al. *Microbiome* study](https://github.com/jfmeadow/Meadow_etal_Surfaces/) * [Westcott & Schloss *mSphere* study](https://github.com/SchlossLab/Westcott_OptiClust_mSphere_2017/) * Questions * What do you notice about the structure of the two projects? * How long did it take to find the code for Figure 1? * Where is the main document? * Where would the data go? ??? So let's compare two projects that we've already looked at and think about the file organization, so we're going to look at the example from Meadow et. al,. the Microbiome, and the article from Sara Westcott and myself that was published in mSphere. And so as we look at these, we want to ask ourselves what do we notice about the structure of the two projects? How long does it take us to find the code for Figure 1? Where is the main document and where would the data go? So I'm going to open these two repositories in separate windows. And so here's the GitHub repository for Meadow et. al., and here's the repository for Westcott and Schloss, okay? So what do you notice about the structure? Well, Meadow has a directory for figure, he also has something for LillisSurfaces_cache/html, not really sure what that's about. Figure, I assume, is going to have the figures, okay? And so here are some of the figures that were in the Meadow et. al. paper, okay? So that's nice, that's good, but everything else is kind of dumped in here into a single directory. He's got code, they've got text files, they've got data all in one directory. Now, I don't want to bust their chops too bad, this is a pretty small project, right? There's maybe 10 files here, there's no need to have a bunch of file structure, directory structure for a small number of files. If you look at the Westcott and Schloss directories, you'll see that there's a directory for code, for data, results, submission. There's a LICENSE, Makefile, README file, and you could then also look at this overview diagram in the readme file as we talked about last time to figure out where different things are. So the Westcott and Schloss is more complicated, it's perhaps a bigger set, bigger or more data sets, and so it needs that structure, whereas the Meadow et. al. perhaps doesn't so much. So if we want to find the code for Figure 1 in Meadow et. al., I think we did this yesterday, we might say, "Well, let's look in functions.R." And if we scroll through here, nothing really jumps out at me as saying it's Figure 1, okay? And maybe it would help first to know what Figure 1 is, so if we look at figure and if we assume this is Figure 1, this is an ordination where they've got different colors for different types of samples and they also have different sized points and I forget from the paper whether or not the size of the points matters. Let's look at another file here, let's look at this LillisSurfaces.Rmd file. So here's the R Markdown file from Meadow et. al. and their surfaces, and if we scroll through here, it looks promising that this will have the information we need to generate Figure 1. And so, let's see. Keep scrolling. We're looking for something that looks like it might be making an ordination or plotting an ordination. "Create an ordination of these sources combined with surface samples," okay. And so, there's a PCA command to build the PCA and to plot the results, okay? So we're not going to go into what does each line of code do, but needless to say, here's the code I suspect for generating Figure 1, okay? And so, it took a little bit of digging but we found it, it's here. One of the nice things about the R Markdown is, it allows you to embed code in with text. If we look now at the Westcott and Schloss repository, let's see if we can find Figure 1 here, okay? And so, if we go code, there's a bunch of files here, a lot of scripts. Do we find anything here for Figure 1? Well, there's build various figures, I suspect it's one of these five figures but I'm not sure where, so let's go back out here. I remember from this documentation earlier, they said if we wanted to build the paper, we'd write "make write.paper," and so I know that "make" means that it uses the makefile. And so if I scroll through this, this is...I will talk about makefiles at a future tutorial, but these are all the instructions for building all of the data sets, and so if we scroll through here, we might look for something that says, "Figure 1," and see what code is used to build Figure 1. So I'm kind of scrolling down through here. "Build figures," okay, well, this tells me how to build the performance figure and I'm not sure if that's Figure 1 or not. I suppose what I could do is to open up results/figures/performance, let's do that. So if we do "results," "figures," it was performance.png was the first one, and this is the first figure from that paper, okay? So performance.png was the file that we want. And so, again, "code," build_performance_figure.R is here, and so here now is the code for building the performance figure, okay? And so one of the things you might notice here is that there are no comments, bad Pat, and so...but we have found the code for Figure 1 for both studies. So where is the main document? If we go back to Meadow et. al., I believe that this is the main document, this .Rmd file, LillisSurfaces.Rmd is the main document, and here, what they're kind of providing is a notebook for how they made the figures and how they did their analysis. For the Westcott and Schloss, if we come back to the home directory for the repository, we might go to "submission" and we see then there's Westcott_OptiClust_mSphere_2016.Rmd and this is more than likely the main document, this is the manuscript that was submitted and that if you...as we learn about literate programming in a future tutorial, we'll see how this code is used to fill in values and plots for generating the final document, okay? So, again, it didn't take very long to find the main document for both repositories. And then the next question is, "Where would the data go?" Where would the data go for Meadow et. al. surfaces? Here it appears that it comes in here, there's this "sourceHabitatsBlastClass," I'm not sure what that is. This is not a...CSV is a pretty generic file format so I'm not exactly sure what's going on, but this is perhaps saying the source and then the abundance and taxonomic ID. They also have a file called RData, which is a way to save data from an R session. That's commonly used. But I don't immediately see how I could add new data to this project if I had other data from other surfaces that I might want to add to compare to...say, you know, if I had data from a university classroom at University of Michigan, how would it compare to theirs? It's not immediately clear how I would do that, albeit, you know, their purposes here are mainly to describe how they did their analysis, not to describe how you would interact with their analysis. And then if I look here in Westcott OptiClust, if I look on "data," the raw data I believe are in here in, like, the "human," "processed," "references,"and "soil". And so, here is the soil metadata. --- ## An aside about large files * GitHub cannot easily handle large datasets * Each file is limited to 100 MB and a repository is limited to 1 GB * Generally only post the data files critical to an analysis * Scripts should document where data goes and how it gets there * We'll talk more about git/GitHub later ??? And so what we notice is missing from this and also the Meadow et. al. data set is that if we look in these, this project analyzed sequence data but there's no sequence data in here and so something we might ask is, "Well, why don't we see those files in here? Why aren't the reference files in here?" We talked about that issue of how we deal with large files. So GitHub cannot easily handle large datasets. Each file has a limit and each file is capped to 100 megabytes and each repository is capped at 1 gigabyte, so, many of our datasets, we cannot host on GitHub. And so we have to do some tricks to get around that, and mainly we need to provide instructions on how to get those files and how to generate intermediate files, we need to provide those instructions with our code and that's part of what we're going to be doing today as we set up a new project. And so, again, we generally only post the data files that are critical to the analysis, things like our metadata and our scripts should document where data go and how it gets there and we'll talk more about using Git and GitHub later for handling large datasets. --- ## General structure for a microbiome study ``` project |- README.md # the top level description of content (this doc) |- CONTRIBUTING.md # instructions for how to contribute to your project |- LICENSE.md # the license for this project |- CITATION.md # instructions on how to cite the project | |- submission/ | |- study.Rmd # executable Rmarkdown for this study, if applicable | |- study.md # Markdown (GitHub) version of the *.Rmd file | |- study.tex # TeX version of *.Rmd file | |- study.pdf # PDF version of *.Rmd file | |- header.tex # LaTeX header file to format pdf version of manuscript | |- references.bib # BibTeX formatted references | |- XXXX.csl # csl file to format references for journal XXX | |- data # raw and primary data, are not changed once created | |- references/ # reference files to be used in analysis | |- raw/ # raw data, will not be altered | |- mothur/ # mothur processed data | +- process/ # cleaned data, will not be altered once created; | # will be committed to repo | |- code/ # any code | |- results # all output from workflows and analyses | |- tables/ # text version of tables to be rendered with kable in R | |- figures/ # graphs, likely designated for manuscript figures | +- pictures/ # diagrams, images, and other non-graph graphics | |- exploratory/ # exploratory data analysis for study | |- notebook/ # preliminary analyses | +- scratch/ # temporary files that can be safely deleted or lost | +- Makefile # executable Makefile for this study, if applicable ``` ??? So we talked about the Noble organization and as I've been doing a lot of projects with microbiome data in my lab, we've come up with a system of organizing our projects that works pretty well for us. And so, this is the overall structure that you saw on the Westcott and Schloss repository where we have the home directory for our project, we have that readme file, maybe a file about contributing, a license, citation, how we want to cite the project. We have a directory for all the files for submission, directory for all of our data, so maybe we have a references directory, a raw directory, a directory that contains output from Mothur. Or if you're using Chime or some other program, you could have a Chime directory. You might also have a directory for processed data, so clean data that won't be altered. You might have a directory for code, also a directory with different results whether they're tables, figures, or pictures, and then you might also have an exploratory directory where you're trying out some different types of analysis and you want to keep track of those in perhaps some type of research notebook, computational research notebook. And then finally, an executable makefile that helps to run the overall project, okay? And so, hopefully, you can look at this structure and agree with me that it makes sense that...and you could come into this without knowing much about the project and figure out where things are. --- ## Bad project: Introduction .center[ <iframe width="570" height="320" src="https://www.youtube.com/embed/Fl4L4M8m4d0?ecver=1" frameborder="0" allowfullscreen></iframe> ] * You have inherited a project from a previous student in the lab * You need to organize it into a logical structure ??? And so I'd like you to participate in a project with me that I'm calling the Bad Project, and if you haven't seen this Lady Gaga parody from the Zheng Lab at Baylor, it's pretty awesome. We can all relate to inheriting a bad project or perhaps starting our own bad project, but you can imagine that perhaps there was a former student or a former postdoc who left and you've now inherited their project and it's up to you to make heads or tails out of what they've done and to turn that into a manuscript. And you look at it and it's just a disaster, they've got kind of the garbage can organization where they've dumped everything into a single directory and you've just watched this tutorial and now you want to organize it into something logical. --- ## Bad project: Exercise * Download the [data](https://github.com/riffomonas/bad_project/archive/v0.1.zip) to a new directory `~/bad_project` ```{bash} wget https://github.com/riffomonas/bad_project/archive/v0.1.zip unzip v0.1.zip rm v0.1.zip mv bad_project-0.1 bad_project cd bad_project ``` * Using the recommended directory structure, move files to the appropriate directories * Use `mkdir new_directory` to create a new directory * Use `mv current_file directory/` to move `current_file` into `directory/` * Don't forget the `/` after `directory` .footnote[Now would be a good time to [log into](../hpc/) your AWS instance or your local HPC] ??? And so what we'll do is, we'll download a data set into a new directory on our EC2 Instance that we'll call "bad_project," and we're going to use the recommended directory structure to move files to the appropriate directories. And so we can...some hints, we can use the command mkdir to create a new directory and we can use mv to move a file from the root directory into our new directory, where the word "directory" here is the name of the directory and "current file" is the name of the file that we want to move, okay? Also don't forget to include the slash after directory because...after the directory name because that slash tells you that it's a directory, okay? So let's go ahead and do this and I'll help you get set up. So we're going to return to our tmux instance... and we're going to type wget https...you can also copy and paste, github.com. Okay. So when we run this, we want to make sure that we're on our EC2 Instance, it's best to get into the practice of using that rather than using your own computer. So if we run that and now we type ls, we see that this archive is in our home directory and we know that it's the home directory because there's a tilde here. Similarly, if we type "pwd," we see that we're in home/ubuntu, your username is ubuntu on this instance, okay? So we can then decompress this archive by typing "unzip v0.1.zip" and this explodes it out. If we type ls, we now see that we've got bad_project-0.1 and then we still have that archive in the home directory. Let's go ahead and get rid of that archive and we can do rm v0.1.zip and let's change the name of our bad project directory from...to get rid of the -0.1, so move bad_project to bad_project, okay? So now if we cd into bad_project, type ls, we see a whole bunch of files. One of the things with tmux that you might notice is if you scroll up, you lose a lot of the output, okay? --- ## General structure for a microbiome study ``` project |- README.md # the top level description of content (this doc) |- CONTRIBUTING.md # instructions for how to contribute to your project |- LICENSE.md # the license for this project |- CITATION.md # instructions on how to cite the project | |- submission/ | |- study.Rmd # executable Rmarkdown for this study, if applicable | |- study.md # Markdown (GitHub) version of the *.Rmd file | |- study.tex # TeX version of *.Rmd file | |- study.pdf # PDF version of *.Rmd file | |- header.tex # LaTeX header file to format pdf version of manuscript | |- references.bib # BibTeX formatted references | |- XXXX.csl # csl file to format references for journal XXX | |- data # raw and primary data, are not changed once created | |- references/ # reference files to be used in analysis | |- raw/ # raw data, will not be altered | |- mothur/ # mothur processed data | +- process/ # cleaned data, will not be altered once created; | # will be committed to repo | |- code/ # any code | |- results # all output from workflows and analyses | |- tables/ # text version of tables to be rendered with kable in R | |- figures/ # graphs, likely designated for manuscript figures | +- pictures/ # diagrams, images, and other non-graph graphics | |- exploratory/ # exploratory data analysis for study | |- notebook/ # preliminary analyses | +- scratch/ # temporary files that can be safely deleted or lost | +- Makefile # executable Makefile for this study, if applicable ``` ??? So what we'd like to do now and what I'm going to shut up and let you do this now is to create the directory structure that we talked about in the...for a microbiome analysis. And so, if you come back to this general structure for a microbiome study, you'll see that, well, we probably need a directory for submission, data, data references, data raw, data mothur, data process, code, results, exploratory, Makefile and that we want to create that directory structure and move our files into those directories, okay? So, again, I'll just briefly show you something that we could do...mkdir code, so I have a code directory and I'm going to move plot_nmds.R into code and if we look at ls code, we see now that code, the directory code, has plot_nmds.R in it, okay? So I will tell you to now pause the video and go off and on your own, give this project some organization. Great, so hopefully, you have something like this. I did add a little Easter Egg in here that if you type "cat solution," you then see my organization, okay, and you can see where I put, say, these fastq.gz files in a data/raw or the HMP_MOCK.fasta in the data references. This is not meant to be the perfect way to do it. This structure is a structure that my research group has found works pretty well for us. Every project is a little bit different, and so it's a starting point to have this structure that helps us to organize our projects. It's not meant to be a straitjacket to force you to do things a specific way, it's a tool to help you to organize things. And the key, again, is that somebody that comes into your home directory, that comes into bad project, should know where they can find different things or they should know what's in data references, okay? And so, if we go from that big garbage can of data to type ls now, and we can see there's a directory called code, data, results, submission, there's a couple of files, LICENSE, Makefile, and README, that it's much easier to get a sense of where things are. So like I said, this is a structure that works really well for my lab and it's a structure that's become fairly standard for my research group. --- ## Bad project: Evaluation * Compare your results to your neighbor * Once you are done, run `cat .solution` to see my results * What do you notice? Do you agree with my organization? --- ## Using a project template * The structure of most microbiome analysis projects is fairly standard * To help with the structure, we created a template, [`new_project`](https://github.com/SchlossLab/new_project/releases/latest) * Return to y our home (~/) directory * Download the zip file for the latest release of `new_project` ??? And so, to help with this structure and because it gets kind of redundant to do the same thing over and over again, I've created a template that I call "new_project," and so we can download new_project which is at that link, we can return to our home directory and we can then download that zip file, decompress it, and use that for the latest release of new_project to start our own analysis. --- ## Exercise .left-column[ * We are going to attempt to generate the paragraph and NMDS plot from [Figure 4](http://aem.asm.org/content/79/17/5112.full) of the Kozich AEM study * Rename the directory `Kozich_ReAnalysis_AEM_2013` * We'll set up the project by following along with the instructions in the [`INSTRUCTIONS`](https://github.com/SchlossLab/new_project/blob/master/INSTRUCTIONS.md) file ] .right-column[ .center[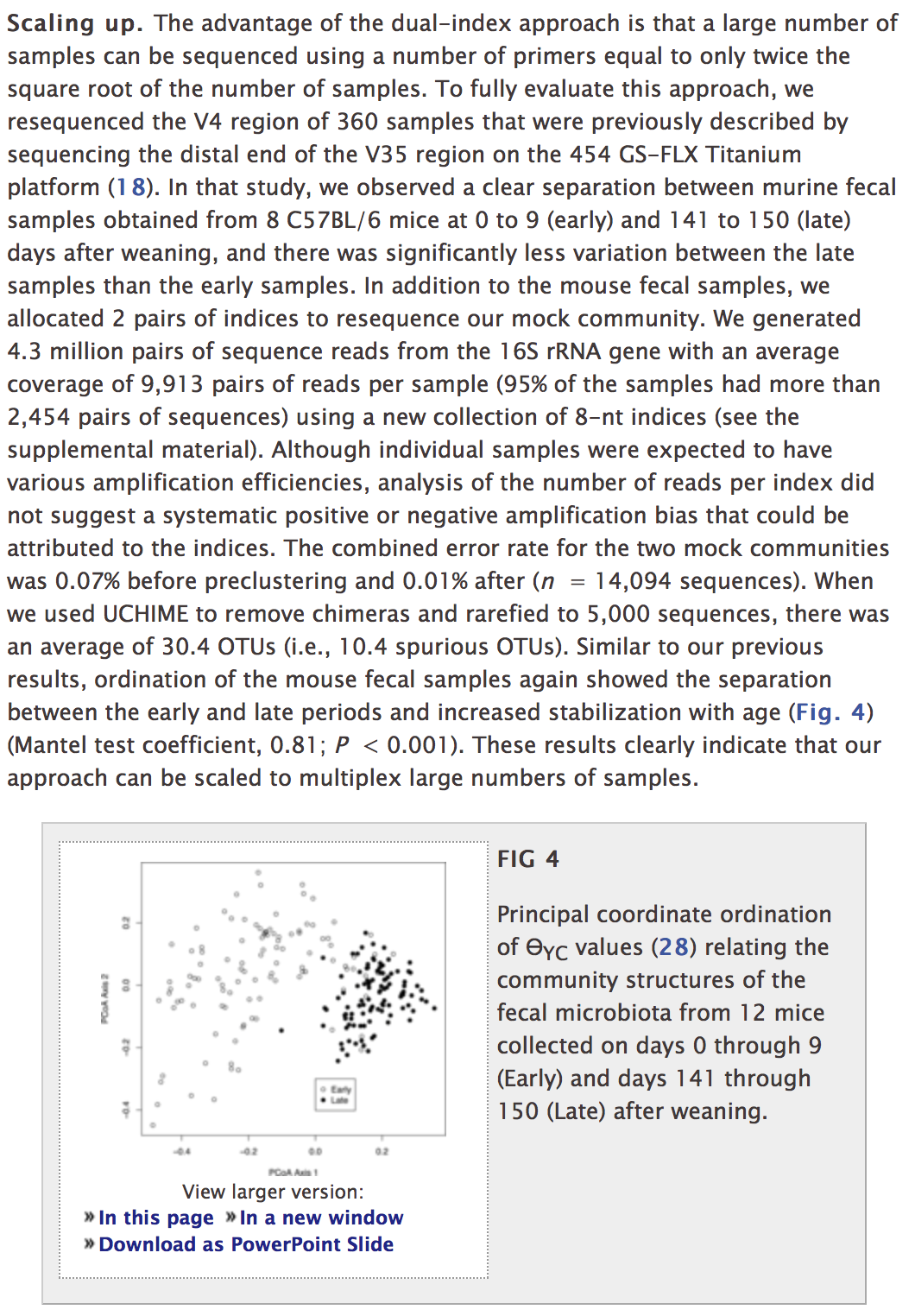 ]] ??? So what we're going to work on in this tutorial as well as in subsequent tutorials is trying to regenerate a paragraph and NMDS plot from the Kozich study that was published in AEM. And so, if you're not familiar with that paper, you might go back and look at it. It's the paper where we describe our method of sequencing 16S genes using Illumina's MiSeq, okay? And so this was published in AEM in 2013. So something I like to do is to name my directories or name my repositories to include the last name of the first author, a very brief, like, one-word, two-word description of the paper, the journal it was published in, and then the year. All right, so I'm going to go ahead and click on this instructions link and go to this tab and here are instructions on how to use the template, so we're going to download the latest release to the directory and decompress, okay? And so if I click on this in a new tab, I see that the latest release is here and there's a link here for a source code that if I right-click on that to copy link, I can say... Well, I need to go back to my home directory so I can do "cd ~" to get back home and I'm going to clear my screen by doing Ctrl+L, then I can type wget and then it downloads it and if I type ls, now I see the 0.11.zip directory which is what...or the archive which I just downloaded as well as my bad_project directory. I now want to unzip this directory and do unzip 0.11.zip, a whole bunch of stuff spits out and if I say ls, I now see I have that archive, bad_project, and new_project. I'll remove the archive and I will move, rename new_project to be Kozich_ReAnalysis, "Reanalysis," can't spell, AEM_2013, then I will cd into the Kozich directory and type ls and we see the structure that we've been talking about. There's a few extra files in here, there's the instructions which we're looking at on the GitHub site and then there is a license for a new project and there's a license for...there's a license for this template, for new_project template. So I'm going to go back to the instructions here on the website just because it's a little bit easier to read. We've already done this where we've renamed the directory to be Kozich_ReAnalysis_AEM_2013. --- ## Modify README.md * Open the README.md document in `nano` * Remove the first line, which starts "Download... " * Edit the following lines as you see appropriate: ```sh ## TITLE OF YOUR PAPER GOES HERE YOUR PAPER'S ABSTRACT GOES HERE ``` * We'll leave the rest as it is for now ??? It tells us then to open the README document in an editor, change the first line to reflect the title of your research study, and the content from that section to the end. You can, but are not obligated to keep the Acknowledgement section, you should keep the directory tree. So I'll do nano README.md, I'm going to get rid of this line. In Nano, you can type Ctrl+K to delete a line and I'm going to replace this with something like "Kozich Re-Analysis Project" and I'll save the abstract for later. This is the overview that we've talked about, I'm going to leave this now but I might want to make sure that I update that as I update my directory structure. And then here are the dependencies, how to regenerate the repository, running the analysis. Okay, so there's other stuff in here that you can update and customize for your own purposes. Do you remember how to save this? All right, so we do Ctrl+O, Ctrl+Enter, Ctrl+X to get out, okay? And if we look back at our instructions, we want to replace the license from the templates repository with the license for your project. --- ## Changing Filenames * At the terminal do the following... ```bash mv newproject_LICENSE.md LICENSE.md ``` * This replaces the license ([CC0](https://creativecommons.org/share-your-work/public-domain/cc0/)) of the `new_project` repository with a license for your analysis ([MIT](https://en.wikipedia.org/wiki/MIT_License)) * Using `nano`, go ahead and read and edit the license to include your name and the year (no need to keep my name unless you are in my lab) ??? So if we look at nano LICENSE, this is a public domain license for the repository, for the template that we made, and that template was part of...have been put into the public domain but your project isn't in the public domain. So we're going to open up nano newproject_LICENSE.md, and see that this is under an MIT license. We'll leave this for now, in a subsequent tutorial, we'll come back and we'll talk about the license again. But we want to make this new project license the real license for the project, so we'll do move, mv newproject_LICENSE LICENSE.md, and that's great. We do ls, we now see that we only have one license directory in here. --- ## Getting ready for `git` * At the terminal do the following: ```bash git init ./ git add . git commit -m "Initial commit" ``` * This is putting your new project under version control using `git` ??? What we'd like to do is to have this directory for our project be under version control. And so to do that, we're going to use Git without really knowing a whole lot about Git, but that'll be okay, and as I said in the introduction, we're going to be talking about Git in the next tutorial as well. So to get started, we're going to type "git init ." I'm going to go ahead and do Ctrl+L to get a clear screen, "git init ." there's spaces between git init and init and the period. We hit Enter, we can then do git add . and then git commit -m "initial commit." --- ## Connecting with GitHub * Go to your home page on GitHub * Create a new repository as you did before with the paper airplane example * This time... * Give the repository the same name as your project (i.e. `Kozich_ReAnalysis_AEM_2013`). * Keep the repository public * Don't initialize with a `README` or `.gitignore` files or a license (you've already got those) * Click "Create Repository" ??? So don't worry about what these three commands did, we'll talk about those more in our next tutorial. So we'd now like to connect this to GitHub, so I'm going to go ahead and go to my profile in GitHub and I am going to create a new repository. --- class: center middle 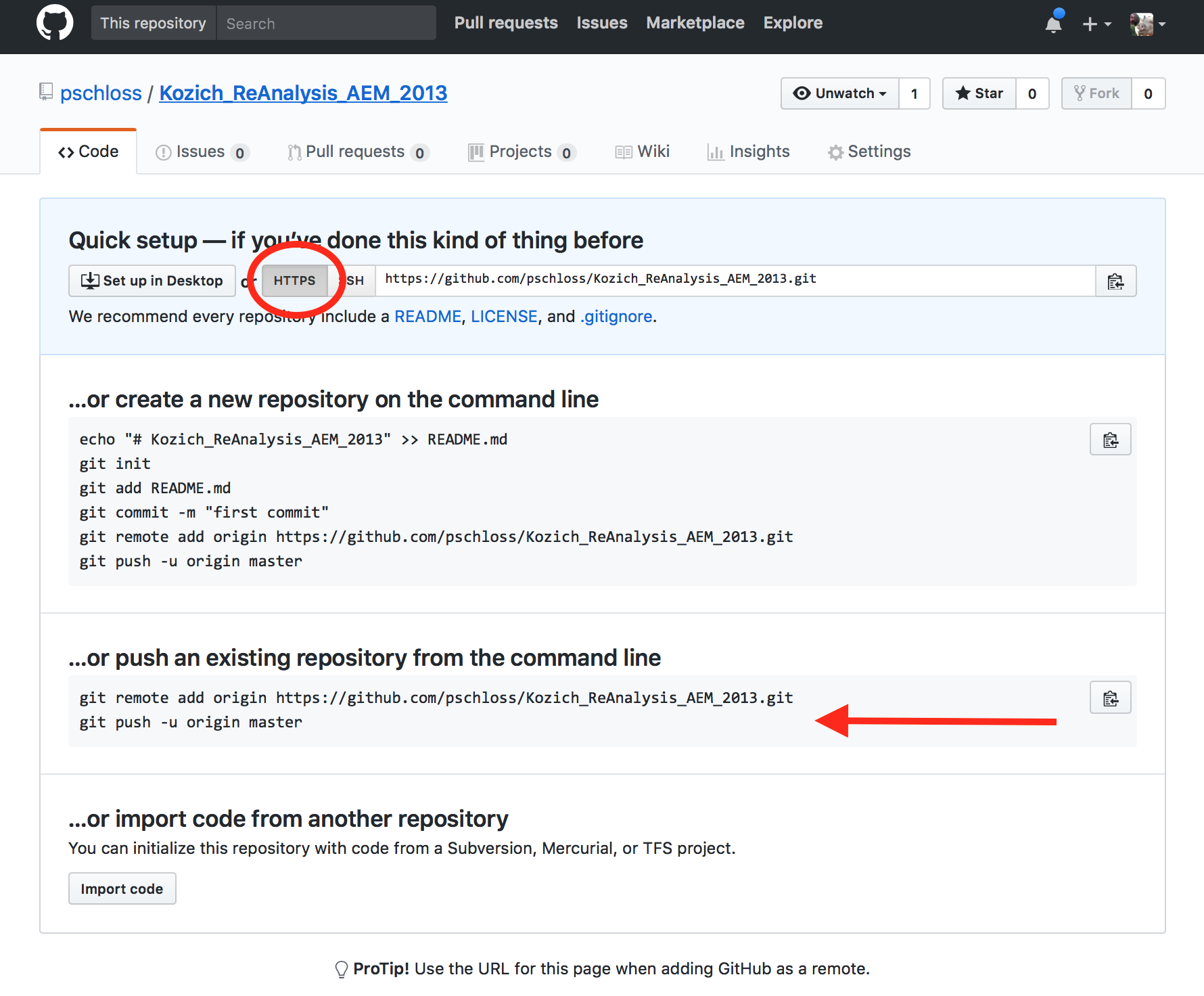 ??? So I'll click on this plus sign up at the top of the screen and do "New repository," and I'm going to name my repository as Kozich_ReAnalysis_AEM_2013. So this repository name does not need to be the same name as your directory over here, it's just kind of helpful to make things align. So we'll go ahead and keep this as a public repository, we don't want to initialize with the Readme or a License because we already have those. Click "Create repository." We already have an existing repository so we want to push an existing repository from the command line. They have a nice little "Copy to clipboard" button here, so I copy that and then over here, I can paste, hit Enter. --- ## Connecting with GitHub * Copy and paste into the terminal the two lines that were highlighted with the red arrow on the previous slide ```bash Kozich_ReAnalysis_AEM_2013 $ git remote add origin https://github.com/pschloss/Kozich_ReAnalysis_AEM_2013.git Kozich_ReAnalysis_AEM_2013 $ git push -u origin master Counting objects: 26, done. Delta compression using up to 8 threads. Compressing objects: 100% (24/24), done. Writing objects: 100% (26/26), 43.09 KiB | 0 bytes/s, done. Total 26 (delta 2), reused 0 (delta 0) remote: Resolving deltas: 100% (2/2), done. To github.com:pschloss/Kozich_ReAnalysis_AEM_2013.git * [new branch] master -> master Branch master set up to track remote branch master from origin. ``` * Back in the browser, refresh the screen ??? Asks for my username, I can say, "pschloss," my password. And so I'm entering my GitHub password and username, okay? So I have now pushed...we'll talk more about that in the next tutorial but I have now pushed the content from this repository up to GitHub. --- class: center middle  ??? If I come back to GitHub and hit Refresh, I now see that I've made a new repository. So this is a bit more of a sophisticated version of a repository than the one we made for our paper airplanes. --- class: center middle  ??? There you go, high-five, good job. But you're not done yet. I have an exercise for you to engage in, to go ahead and to get the raw data and the reference files that we're going to need to run this analysis. --- ## Cleaning up * Remove the instructions file and commit ```bash git rm INSTRUCTIONS.md git commit -m "Remove instructions document" git push ``` * Refresh your GitHub page and make sure the `INSTRUCTIONS.md` file goes away * If you ever need the information in the `INSTRUCTIONS.md` file, you can retreive it from the `new_project` [repository](https://github.com/SchlossLab/new_project/blob/master/INSTRUCTIONS.md) --- ## Let's go! * You need... * A partner (preferably someone with mothur experience) * Familiarity with [the original paper](http://aem.asm.org/content/79/17/5112.full) * The data (hint: grab the data from the "With genomes/metagenomes" link) * The reference files * mothur * Dump the text for the figure lengend and the paragraph titled, "Scaling up", in the main REAME.md * Obtain the files and put them in the correct locations * Do your best to document what you are doing ??? So, ideally, you'd work with a partner but I realize that you might be working individually so that's fine. Hopefully, you are working with somebody that has some Mothur experience. This is not going to be a Mothur tutorial series, we will not use much Mothur at all in here or talk much about Mothur, so it's not super-critical. It would be good for you to be familiar with the original manuscript, with the original paper, but again, it is important. So to help you find that, if we go to...click on this in a new tab, this will open up at the AEM website. So this is the manuscript, and so you're going to need to look at this to figure out what kind of references did I use? Where are the raw data? I was not a good kid, I did not put them in the SRA at the time. And so you're going to want to look through here and to make some judgments about where the data are and perhaps what references you need and what software you need. So what I'd like you to do then is that you need to get the data, and as my hint, at the website you'll go to there will be a link for "With genome/metagenomes," you want those data, you'll want to get the reference files, you'll want to get Mothur, and you'll also want to take the text for the figure legend and the paragraph titled, "Scaling Up," in the README, so the figure legend for Figure 4. And so you're going to want to obtain the files and put them in the correct locations in your EC2 Instance and I'd like you to do your best to document what you are doing. So if I repeat everything we've done with this New Project template and I want to get going to create this Kozich re-analysis following your approach, I need instructions from you as to what you did. --- ## Helpers * `wget` - pulls down large data files to the current directory (or `curl -LO`) * `tar xvf` - unpacks a `.tar` file * `tar xvzf` - unpacks a `.tar.gz` or `.tgz` file * `rm` - removes files * In a mardown file you can have text rendered as code by putting the code inside of two sets of three back ticks ``` ``` code/mothur/mothur -v `````` On GitHub, this will be rendered as... ```bash code/mothur/mothur -v ``` ??? As some helpers, we've already used a tool called wget. Wget is useful for pulling down large data files to the current directory. There's another tool called curl, I learned wget so I like wget. tar xvf will unpack a .tar file, tar xvzf unpacks a tar.gz or a tgz file. As we've already seen, rm will remove files or delete them. And if you're making a Markdown file like a readme file, you can have text rendered as code on GitHub by putting the code inside of two sets of three backticks. So this is a backtick which is the lower character on the key with a tilde, so it's the key right above the Tab on the left side of your keyboard, okay? --- ## When you're done... * .alert[Wait!] Do not commit or push your changes * Add me/your PI as a collaborator * "Settings" -> "Collaborators" -> Enter handle -> "Add collaborator" * I'm at `pschloss` ??? So when you're done, wait, don't do...don't get fancy and make a GitHub commit or push, don't do that. We'll do that in the next tutorial and please wait for me. If you'd like, add me or your PI as a collaborator to this project and you can do that by going to "Settings," "Collaborators," and then enter the handle, so mine is pschloss, which is me, but that's kind of silly because I'm already collaborating with myself. So if you want to add me or if you want to add your PI or somebody else, that's great. So look at this list of things that you need to do and go ahead and do them, stop the video, and when you're done, come back and I'll show you what I did. Hopefully, that exercise wasn't too challenging. Hopefully, you're able to get the sequence data, the raw data, the reference files, Mothur, and the text for the paragraph and to dump that into the README file. In addition, I hope you're able to document what you did and the various steps you took to obtain those files. I'm going to share with you what I wrote for my code. And, again, it's not critical that yours matches exactly what mine looks like but what I'm trying to illustrate with my README file, this one here is in the code directory, is how I obtained the Linux version of Mothur. --- `code/README.md` ```bash # README Obtained the Linux version of mothur (v1.39.5) from the mothur GitHub repository ```bash wget --no-check-certificate https://github.com/mothur/mothur/releases/download/v1.39.5/Mothur.linux_64.zip unzip Mothur.linux_64.zip mv mothur code/ rm Mothur.linux_64.zip rm -rf __MACOSX `````` ``` mothur should output the version by doing... ```bash code/mothur/mothur -v `````` ??? And hopefully, you can see from this code here, this Bash code, that this would allow me to copy and paste these lines into a terminal to get these... to get Mothur installed into my code directory. So then if I ran code/mothur/mothur with the -v, I would get the version number. --- `data/raw/README.md` ```bash # README Obtained the raw `fastq.gz` files from https://www.mothur.org/MiSeqDevelopmentData.html * Downloaded https://mothur.s3.us-east-2.amazonaws.com/data/MiSeqDevelopmentData/StabilityWMetaG.tar * Ran the following from the project's root directory ```bash wget --no-check-certificate https://mothur.s3.us-east-2.amazonaws.com/data/MiSeqDevelopmentData/StabilityWMetaG.tar tar xvf StabilityWMetaG.tar -C data/raw/ rm StabilityWMetaG.tar `````` ??? Similarly, here's the instructions to get the fastq.gz files from the website where we posted the data and you can see there's a wget command here, there's a tar call to decompress the tar file, and then we got rid of that tar file because I had put all the data into data raw, okay? So there's many ways that you might get through the desired output of having all your fastq.gz files in data raw, okay? This is how I did it. The key that I want to emphasize is that we can use code and we can use text explanations to describe to somebody how they can achieve what we did and they can see what we did. --- `data/references/README.md` ```bash # README Obtained the silva reference alignment from the mothur website: ```bash wget https://mothur.s3.us-east-2.amazonaws.com/wiki/silva.seed_v123.tgz tar xvzf silva.seed_v123.tgz silva.seed_v123.align silva.seed_v123.tax code/mothur/mothur "#get.lineage(fasta=silva.seed_v123.align, taxonomy=silva.seed_v123.tax, taxon=Bacteria);degap.seqs(fasta=silva.seed_v123.pick.align, processors=8)" mv silva.seed_v123.pick.align data/references/silva.seed.align rm silva.seed_v123.tgz silva.seed_v123.* rm mothur.*.logfile `````` ``` Obtained the RDP reference taxonomy from the mothur website: ```bash wget -N https://mothur.s3.us-east-2.amazonaws.com/wiki/trainset14_032015.pds.tgz tar xvzf trainset14_032015.pds.tgz mv trainset14_032015.pds/trainset* data/references/ rm -rf trainset14_032015.pds rm trainset14_032015.pds.tgz `````` ??? And so if you looked at my three lines here, you could say, "Oh, Pat, you have a bug on Line 2, you need to use this parameter instead of that parameter." And so, it's transparent, then I can see what was going on or somebody else can see what was going on. The advantage of writing it as code with these three lines of wget, tar, rm, is that I could copy and paste these into my terminal to then generate those files and to get the files and put them automatically into data raw without me having to do anything else. That they're instructions for the computer and they're instructions for somebody else that might come in the future. Here are the instructions to get the references that I'll use, I used a silva reference alignment file that I got from the Mothur website, as well as the RDP reference taxonomy that I also got from the Mothur website, okay? So if you struggled a little bit to find these on the Mothur website, again, it's not a big deal. That comes a bit with some of the familiarity with Mothur, and again, as I'm saying this, I'm realizing I'm assuming that somebody knows how to use Mothur and where they can get these reference files. And so, again, if people already had this code and could see it in my repository, they would know exactly where I got the trainset version 14 from this code in my README file but that wasn't immediately obvious from looking at the manuscript. --- `README.md` ```bash ## Re-analysis of Kozich dataset **Scaling up.** The advantage of the dual-index approach is that a large number of samples can be sequenced using a number of primers equal to only twice the square root of the number of samples. To fully evaluate this approach, we resequenced the V4 region of 360 samples that were previously described by sequencing the distal end of the V35 region on the 454 GS-FLX Titanium platform (18). In that study, we observed a clear separation between murine fecal samples obtained from 8 C57BL/6 mice at 0 to 9 (early) and 141 to 150 (late) days after weaning, and there was significantly less variation between the late samples than the early samples. In addition to the mouse fecal samples, we allocated 2 pairs of indices to resequence our mock community. We generated 4.3 million pairs of sequence reads from the 16S rRNA gene with an average coverage of 9,913 pairs of reads per sample (95% of the samples had more than 2,454 pairs of sequences) using a new collection of 8-nt indices (see the supplemental material). Although individual samples were expected to have various amplification efficiencies, analysis of the number of reads per index did not suggest a systematic positive or negative amplification bias that could be attributed to the indices. The combined error rate for the two mock communities was 0.07% before preclustering and 0.01% after (n = 14,094 sequences). When we used UCHIME to remove chimeras and rarefied to 5,000 sequences, there was an average of 30.4 OTUs (i.e., 10.4 spurious OTUs). Similar to our previous results, ordination of the mouse fecal samples again showed the separation between the early and late periods and increased stabilization with age (Fig. 4) (Mantel test coefficient, 0.81; P < 0.001). These results clearly indicate that our approach can be scaled to multiplex large numbers of samples. ### Overview project |- README # the top level description of content (this doc) <snip> ``` ??? And then finally, I took that paragraph "Scaling Up" from the Kozich paper and put that into my README.md direct file in the home directory of my project. So, again, don't feel like you have to use the exact same approach I used to documenting this and using the Bash code to get the files and to put the files where they belong, the goal is to get the files, put them where they belong and by whatever means necessary, but hopefully, those means are reproducible and that somebody else could come along and do it as well. And so something you might do is try deleting all of those files that you just downloaded and rerun your instructions and see if you can do it without getting any error messages. So finally, I'd like to leave you with some exercises to work on. I don't want you to enter these into your AWS Instance but think about and write out perhaps by hand or in a document the commands to create a new directory, how to move from your home directory to some other directory, how to create a new Git repository, how do you get data from an internet-based resource, how do you move a file from one directory to another, how do you rename a file, and then how do you decompress a file ending in zip, tar.gz, and tar. And if you can do all of these things, then you'll be in really good shape for the future tutorials. Before we close, we want to remember that we need to close our instance, and so we need to come to our shell and type "exit." That will get us out of our tmux window and if we type "exit" again, we're now at our home directory, and if I type "exit" again, that window will close and if I come back to my EC2 management console, I can do "Actions," "Instant State," "Stop," and I'll say, "Yes, stop." --- ## Write the command line code that does the following... * Creates a new directory called `my_awesome_directory` * Move from your home directory to the `my_awesome_directory` directory * Create a new git repository * Get data from a internet-based resource * Move a file from one directory to another * Rename a file * Decompresses a file ending in zip? tar.gz? tar? ??? * mkdir my_awesome_directory * cd ~/my_awesome_directory * git init . * wget --no-check-certificate https://github.com/mothur/mothur/releases/download/v1.39.5/Mothur.linux_64.zip * mv current_file new_directory/ * mv newproject_LICENSE.md LICENSE.md * unzip v0.1.zip; tar xvzf trainset14_032015.pds.tgz; tar xvf StabilityWMetaG.tar -C data/raw/ ??? Well, that was a lot of work. I really hope that you took the time to pause the video and engage in the material to fix the Bad Project and to use the New Project template to organize the data and reference files that we'll need for the Kozich re-analysis effort. I know there's a temptation to watch these videos straight through. I know when I watch other people's videos, I do that myself, but you really get so much more out of the experience by doing the work in parallel with me. Once you feel like you've understood the material and are pretty confident in your skills, take a look back at a project you're working on for your own research and think about how the files are organized. I don't mean to imply that you've got a bad project but perhaps you can replicate that exercise with your own project to improve the organization. You can also go back and generate some Readme files to describe how and where that you got the data and reference files for your own project. So this is the second time that we've worked with Git. The first time was with the paper airplane example. We used Git on GitHub even though you didn't really feel like you are using Git perhaps. In the next tutorial, we'll do a deep dive into Git. That tutorial will give us the skills we need to keep track of the progress of our analysis. Until then, keep practicing and I'll talk to you soon.